DockingServer – Getting Started
This document will guide you through the process of docking and introduce the main features of DockingServer.
My Proteins
You can set up your protein first using this page. You can use your own (*.pdb format) file and upload a protein or you can download the desired protein from the RCSB protein data bank.
If you choose the latter option, type in either the code or the name of the protein then click on the "Search" button. With that you can see the details of protein including the code, compound, release date, source, author list a resolution. If the wanted protein is on the list you can choose it checking the box next to it then click on the "Download" button. You can choose a folder for the protein or you can create a new folder for it. If you did not find the protein you can start a new search.
Once the protein is in the "My Proteins" folder you can set up the properties clicking on the name of the protein.
The first step is Protein clean.
Here you can select the protein chains that you want to include in your simulation. You can select a chain clicking on the name of it. Once the name is highlighted you selected the chain. Note that you can select more than one chain by holding the "Ctrl" key and clicking on the name of the wanted chains at the same time. If you want to cancel the selection and select another chain you should click on the "clear selection" text below.
You can find the co-crystallyzed ligands of the protein and select heteroatoms and water molecules. However, please note: do not select ligands, if you are going to dock the same binding site. If you are done with these settings click on the "Confirm" button.
During the next step you need to select simulation box. You can use the default values or initialize the size of the box and give parameters in Angstrom as well. For the easier work DockingServer visualize the protein in 3D. If you choose the "Hide not selected" option you will see only the amino acids contained by the box. By the "Show Sequence" checkbox you can see the single letter amino acid code sequence also, and choose the center of the box. If you are done with these settings click on the "Confirm" button.
After the setup of the protein the program will compute the parameters. This may take several minutes but during the computation you can continue with setting up your ligand.
My Ligands
You can upload also a ligand using the "Upload Ligand" button, you can draw a ligand (java runtime environment must be installed on your computer) or you can download the desired ligand from the PubChem data bank. The molecule drawing application is easy to use. You can select the wanted atoms on the right side and you can add an atom by drag and drop it with your mouse. You can choose between several bond types (Fig. 1) or can use preset forms also. The Edit – Undo and Redo functions facilitate your work. After you named and drew the ligand you can save it.
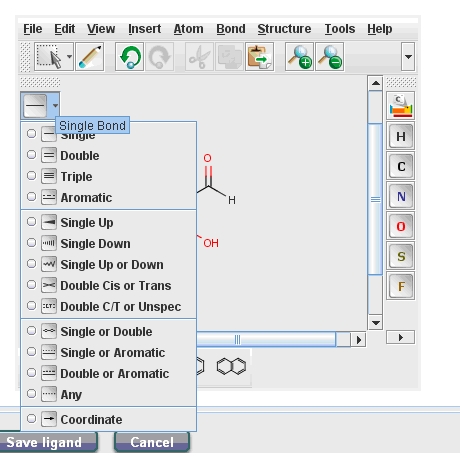
After selecting the ligand you need to set up the Ligand parameters. You can choose the geometry optimization and charge calculation methods, as well as pH.
If you are done with the setup click on the "Confirm" button and the program will prepare your ligand which also can take several minutes. With prepared protein and ligand you are ready to start docking.
My Dockings
This tab contains your past dockings and you can start a new docking here also by clicking on the "Start Docking" button.
First you need to select the wanted ligand(s) then click the next button. (If you do not see the ligand on the list double check to see if the My Ligands tab requires more information and waiting for user input.) and you should select protein(s). On the settings tab you can select the target folder where the actual docking will be saved and ask for e-mail notification upon completion (if you would like to be notified when the docking is done check this field).
You can set up more parameters using the advanced settings. The first three fields are for making changes to the state variables and affect SA (simulated annealing) and the evolutionary methods, GA (genetic algorithm), LGA (Lamarckiam GA) and EP (evolutionary programming). They are defined using the "tstep" (translation step), "qstep" (quaternion step / rigid-body orientation) and "dstep" (torsion step/ dihedral angles) keywords.
The default values are:
- tstep: torsion step (0.2 Å)
- qstep: rigid-body orientation step (5°)
- dstep: dihedral angles step (5°)
- rmstol: the root mean square deviation tolerance (2.0 Å)
- ga_pop_size: the number of individuals in the population. Each individual is a coupling of a genotype and its associated phenotype, usually this number is fixed throughout the run. Typical values range from 50 to 200. (default value: 150)
- ga_num_evals and ga_num_generations: limits for the docking program. AutoDock counts the number of energy evaluations and the number of generations as the docking run proceeds: the run terminates if either limit is reached. (default values are: 25000000 (ga_num_evals) and 540000 (ga_num_generation))
- ga_run: the number of runs to be executed. This command invokes the new hybrid, Lamarckian genetic algorithm search engine, and performs the requested number of dockings. (default value: 100)
Once you are done with the settings click on the "Start" button to let the program start docking. The docking time depends on the given parameters (e.g. ga_run).
Result file formats
You can download the docking results in several file types listed below:
- .jpg files can be associated with any picture viewer program, so you can put the pictures of your results in a demonstration or documentation.
- results.pdf can be associated with Adobe Acrobat Reader and contains the details of your actual docking so you can browse your results offline as well.
- *.map files can be associated with Autodock, and it contains several information about the protein. AutoGrid calculates two different types of carbon grid maps, one for aliphatic carbons in the ligand (*.C.map) and one for aromatic carbons in the ligand (*.A.map).
- The Grid Map Field File has *.maps.fld extension. This is essentially two files in one. It is both an AVS field file, and AutoDock input file with AutoDock-specific information "hidden" from AVS in the comments at the head of the file. AutoDock uses this file to check that all the maps it reads in are compatible with one another and itself.
- The AutoDock Docking Parameter file has *.dpf extension.
- The Protein Data Bank with Partial Changes file has *.pdbq extension.
- PDBQ with Solvation Parameters has *.pdbqs extension.
- The AutoGrid Grid Parameter File has *.gpf extension.
- You can download your docking result in several molecule formats such as hb2, nb2, mol2 and pdb formats. The hb2 and nb2 file format contains information about hydrogen bonds and can be associated and opened with HBPLUS.
Features of Molecular Docking Server
You can
- start single molecular docking calculations or high-throughput virtual screening with few clicks
- carry out focused docking to a known binding site
- carry out blind docking experiments to determine possible binding site(s)
- calculate inhibition constants, binding geometry, secondary interactions & much more
- prepare publication quality figures and a method section for your reports automatically
- organize proteins and ligands into user-defined libraries
- carry out molecular docking on-line
Steps of ligand docking
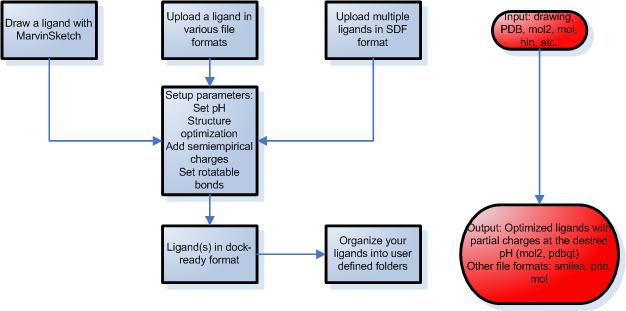
STEP 1 – Preparation of ligands
- Draw your ligands using a Java applet, upload a single ligand file or multiple ligands.
- Draw chemical structures by MarvinSketch, a Java based program with a constantly growing list of editing features and a number of templates to make molecule drawing simpler.
- Upload a ligand in MDL MOL, SYBYL MOL2, PDB, HYPERCHEM HIN or SMILES format.
- Upload multiple ligands in SDF format.
- You can set various parameters during the simulation such as desired pH, structure optimization and partial charge calculations using molecular mechanics or semiempirical quantumchemical methods.
- Set up rotatable bonds and atom types automatically or modify manually.
- Download the attached files in several file formats including mol, pdb, mol2 and pdbqt.
- Organize your ligands into self-defined folders. This way the ligands are saved for later docking calculations.
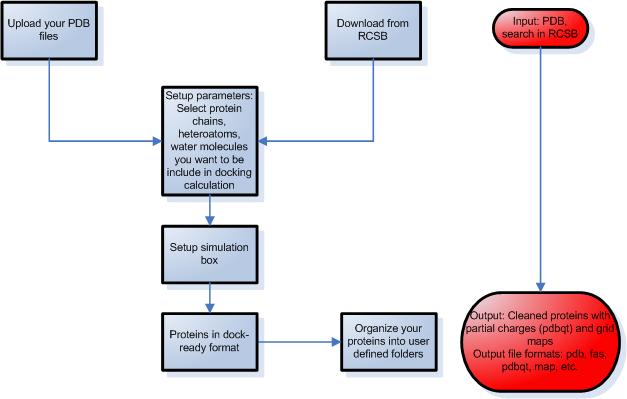
STEP 2 – Preparation of proteins
- Upload protein structures from your files or download them from the Protein Data Bank using Docking Server by providing the entry code or by text search.
- Select the protein chain, heteroatoms, ligands and waters present in the protein pdb file that you want to be included in docking calculation in the process of protein setup.
- Setup the simulation box using one of the following ways:
- select known binding site through a co-crystallized ligand
- select the center of mass of the protein
- select the coordinates of the box center
- select amino acid residues that define the binding site
- Molecular Docking Server calculates necessary map files for each atom type and prepares the input files for docking calculations.
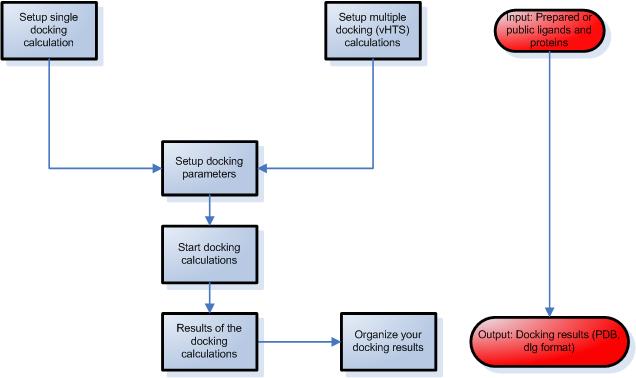
STEP 3 – Setup ligand protein docking calculations
- Select a protein and a ligand from your library.
- Modify advanced parameters during the simulation, such as number of runs, number of evaluations etc.
STEP 4 – Evalution of results
- Choose an image from the image gallery or render in Molecular Docking Server.
- Analyze the secondary interactions between the protein and ligand.
- Create a method section for your reports automatically.
DockingServer Frequently Asked Questions
- What is DockingServer good for?
- Where can I get molecule coordinate files?
- How can I create the PDB file for a particular sequence?
- How do I create high resolution images?
- What are the restrictions in the free version?
- I used DockingServer in my scientific work. How can I cite it?
- What hardware is needed to run dockings by the DockingServer?
- What software is needed to run dockings by the DockingServer?
- What are the hardwares behind the DockingServer?
- What are the softwares behind the DockingServer?
- Are the ligands flexible?
- Can DockingServer handle multiple-ligand files?
- Are the protein sidechains flexible?
- What about protein backbone flexibility?
- Can I dock multiple ligands to the same protein?
- How do I measure distances within DockingServer?
- Is it possible to use Dockingserver for protein-protein docking?
- What is Dockingserver good for?
- Molecular docking is a method which predicts the preferred orientation of one molecule (ligand) to a second (protein) when forming a stable complex. Moreover, knowledge of the preferred orientation may be used to calculate the binding affinity between two molecules (e.g. free enthalpy of binding). With the aim of DockingServer, you do not need to be a computer expert to produce high-quality molecular modeling results. DockingServer does the most difficult parts of the molecular modeling job automatically: it calculates the geometry and electronic properties of the ligands at a given pH, prepares the protein structure to a dock-ready form, runs the calculations, and analyzes the results. Dockingserver can automatically handle multi-ligand files and process thousands of ligands with just a few clicks. Similarly, thousands of docking runs can be done with a few steps to identify the most effective small molecules for given targets.
- Where can I get molecule coordinate files?
- In addition to the public proteins and ligands that are distributed with DockingServer, the major source of data files is the Protein Data Bank at Brookhaven National Labs. This is the main repository for all of the world"s known 3D crystallography and nuclear magnetic resonance (NMR) structures of proteins and nucleic acids. You can download these structures within the DockingServer. Small molecule coordinates are not necessary for Dockingserver, because they can be computed form 2D structures. There are many databases if you need input structures for your ligands, e.g. the ZINC database.
- How can I create the PDB file for a particular sequence?
- If you are interested in a protein, which experimental structure has not been determined yet, its structure should be modeled first. There are several ways to do that: i, You can try homology modeling programs, like Modeller (www.salilab.org/modeller) ii, you might consider using homology modeling servers, e.g. the Swiss-Model server that performs comparative homology molecular modeling to return the potential 3D structure to you as a PDB file iii, or you can contact our experienced team at info@virtuadrug.com to produce high-quality models for your research.
- How do I create high resolution images in DockingServer?
- High-quality images of dockings are automatically produced by DockingServer. If you are not satisfied with the images for any reason, you could download the complex structures and visualize them in any molecular viewer. We recommend using Pymol (pymol.sourceforge.net), which can be used for producing publication quality images of the molecular models.
- What are the restrictions in the free version?
- In short: While our software is free, our hardwares are not. Therefore, we cannot grant processor priority and data storage for free users. Our subscribers get higher priority to run molecular modeling jobs, and their own login name to store and organize their data. Moreover, multi-ligand file processing (SDF) is available only for our subscribers because the high processor utilization.
- I used DockingServer in my scientific work. How can I cite it?
- Bikadi, Z., Hazai, E.
Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock
J. Cheminf. 1, 15 (2009) - What hardware is needed to run dockings by the DockingServer?
- The calculations use our servers, a computer with an ability to run Web browsers and an Internet connection are enough to run calculations.
- What software is needed to run dockings by the DockingServer?
- Molecular modeling and converting softwares run on our servers so you will not need to install any. However, to be able to process the small molecules, you will have to install Java on your computer. It is also recommended that you install a molecular visualisation software, DockingServer is tested to be compatible with Pymol (http://pymol.sourceforge.net).
- What are the hardwares behind the DockingServer?
- Currently DockingServer runs on Dual Quadcore Xeon 2.33GHZ "5345" servers.
- What are the softwares behind the DockingServer?
- Our pipeline software was built on several world-leading applications in the field of molecular modeling. Autodock (http://autodock.scripps.edu), the most popular moleculer docking program is used for molecular docking calculations. Chemaxon tools (www.chemaxon.com) are used for small molecule visualisation and processing. MOPAC2009 (http://openmopac.net) and the revolutionary PM6 semiempirical method can be used to calculate small molecule geometries and electric properties.
- Are the ligands flexible?
- Yes, the ligands are flexible about their single bonds. DockingServer trys to identify the rotatable bonds, which are allowed to rotate freely during the docking runs. If you want to dock rigid ligands, it is possible to change bonds to rigid using the DockingServer.
- Can DockingServer handle multiple-ligand files?
- Yes, Dockingserver is able to automatically convert SDF files to dock-ready form. It is very useful for high-throughput virtual sreening to identify the best compound in small molecule libraries to given targets.
- Are the protein sidechains flexible?
- Currently the whole protein is treated as a rigid molecule. We are intensively working for an easy-to-use method to allow sidechain flexibility, it is expected to be ready within a few months.
- What about protein backbone flexibility?
- Handling protein backbone flexibility during docking calculations is a very difficult task. We are considering the possibilities of how to involve backbone flexibility calculations into the DockingServer.
- Can I dock multiple ligands to the same protein?
- Yes, it is possible. It can be don in subsequent steps: docking the first ligand to the ligand-free protein, then in a second step using the protein with the docked ligand as a target to dock the second ligand.
- How do I measure distances within DockingServer?
- The most important distances for secondary interactions are listed in the docking results section. If you are interested in other distances, you should download the resultant PDB file of the complex structure and measure the distances in a molecule viewer. We recommend using Pymol (pymol.sourceforge.net), which will help you measure distances with ease.
- Is it possible to use Dockingserver for protein-protein docking?
- Currently, protein-protein docking is not possible by DockingServer, however, we are planning to expand the capabilities of Dockingserver to be able to run protein-protein docking jobs.