SO2CH3CH2CH3 to 6cox - OXIDOREDUCTASE
Author: Dr. Wanda Rodriguez Rivera - 14/12/19, 09:54
More docking results of Dr. Wanda Rodriguez Rivera
More docking results of Dr. Wanda Rodriguez Rivera
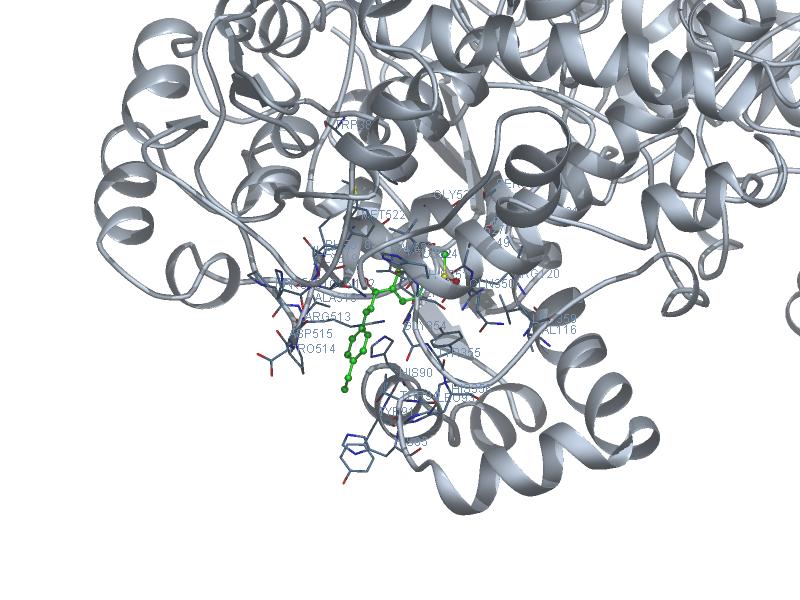
Result table
Rank | Est. Free Energy of Binding | Est. Inhibition Constant, Ki | vdW + Hbond + desolv Energy | Electrostatic Energy | Total Intermolec. Energy | Frequency |
|---|---|---|---|---|---|---|
1 | -7.85 kcal/mol | 1.75 uM | -9.22 kcal/mol | -0.09 kcal/mol | -9.31 kcal/mol | 30 |
Interaction Table
polar | hydrophobic | pi-pi | other | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
| ||||||||||||
|
|
|
| ||||||||||||
|
|
|
| ||||||||||||
|
|
| |||||||||||||
|
|
| |||||||||||||
|
|
| |||||||||||||
|
| ||||||||||||||
|
| ||||||||||||||
|
| ||||||||||||||
| |||||||||||||||
| |||||||||||||||
| |||||||||||||||
| |||||||||||||||
| |||||||||||||||
| |||||||||||||||
| |||||||||||||||
| |||||||||||||||
| |||||||||||||||
| |||||||||||||||
| |||||||||||||||
| |||||||||||||||
| |||||||||||||||
| |||||||||||||||
|

Docking calculations were carried out using DockingServer (Bikadi, Hazai, 2009). Gasteiger partial charges were added to the ligand atoms. Non-polar hydrogen atoms were merged, and rotatable bonds were defined.
Docking calculations were carried out on SO2CH3CH2CH3 protein model. Essential hydrogen atoms, Kollman united atom type charges, and solvation parameters were added with the aid of AutoDock tools (Morris, Goodsell et al., 1998). Affinity (grid) maps of ×× Å grid points and 0.375 Å spacing were generated using the Autogrid program (Morris, Goodsell et al., 1998). AutoDock parameter set- and distance-dependent dielectric functions were used in the calculation of the van der Waals and the electrostatic terms, respectively.
Docking simulations were performed using the Lamarckian genetic algorithm (LGA) and the Solis & Wets local search method (Solis and Wets, 1981). Initial position, orientation, and torsions of the ligand molecules were set randomly. All rotatable torsions were released during docking. Each docking experiment was derived from 100 different runs that were set to terminate after a maximum of 2500000 energy evaluations. The population size was set to 150. During the search, a translational step of 0.2 Å, and quaternion and torsion steps of 5 were applied.
Created with DockingServer
DockingServer Reference
-
Bikadi, Z., Hazai, E.,
Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock
J. Cheminf. 1, 15 (2009)